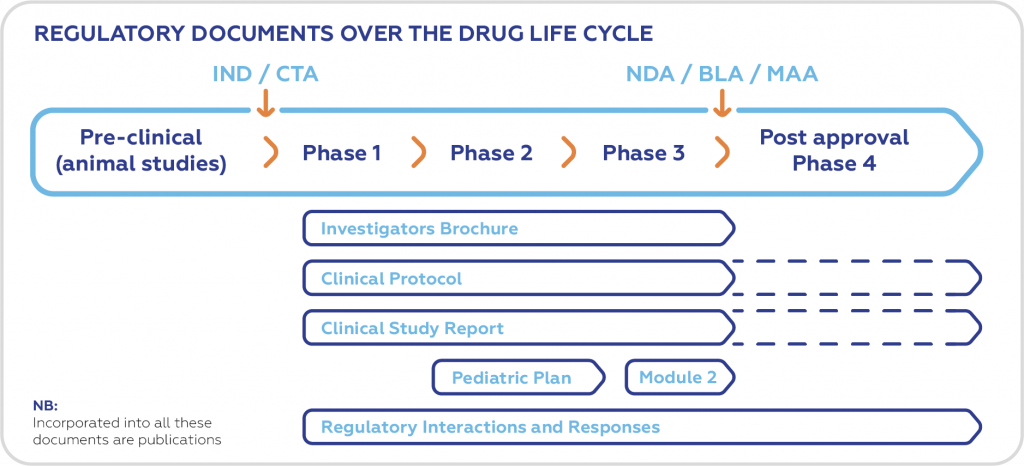
The need for medical writing in the pharmaceutical industry is increasing as companies develop more new drugs and medical devices, and various scientific documents need to be generated for submission to regulatory authorities during their approval process. Regulatory medical writing means creating the – huge – documentation that regulatory agencies require in the approval process for drugs, devices and biologics. All new drugs go through the increasingly complex process of clinical trials and regulatory procedures that lead to market approval. This demand for the clear articulation of medical science drives the demand for well written, standards-compliant documents that the regulators can easily and quickly navigate through (i.e. in Common Technical Document [CTD] format), read and understand. Medical writing as a function has become established especially in the pharmaceutical industry because the industry recognized that it requires special skill to produce well-structured documents that present information clearly and concisely.
A medical writer is often involved in the preparation of the following documents:
Research documents, e.g.:
- Clinical trial protocols
- Investigators′ Brochure
- Informed Consent Documents
- Study reports
- Research proposals
Regulatory Documents, e.g.:
- Summary of Product Characteristics & Patient Information Leaflets
- Clinical study reports
- Subject narratives
- Regulatory submission documents, e.g. Common Technical Document (CTD) modules such as nonclinical and clinical overviews & summaries, expert reports, safety and efficacy summaries
- Safety reports, e.g. Periodic Safety Update Reports (PSURs), bridging reports, periodic benefit-risk evaluation report (PBRER), Development Safety Update Report (DSUR), Risk Management Plans etc.
- Disclosure of clinical results on e.g. clinicaltrials.gov and EudraCT
COURSE CURRICULUM
- Drug Discovery and Clinical Research
- Clinical Study Protocol
- Clinical Protocol Amendments
- Investigator Brochures (IBs)
- Informed Consent Forms (ICFs)
- Case Report Forms (CRFs & e-CRF)
- Source Documents
- Investigational Medicinal Product Dossier (IMPD)
- Patient Safety Narratives
- Narratives for Adverse Drug Reactions & Adverse Events
- Regulatory Documents
- Clinical Study Reports (CSRs)
- Quality Checks QC / Quality Review QR
- US FDA Guidelines
- EU & Other Foreign Regulations
- Global Regulatory Submissions
- New Drug Application (NDA)
- IND Submissions
- Abbreviated New Drug Application (ANDA)
- Independent Ethics Committee (IEC)
- Standard Operating Procedures (SOPs)
- ICH/GCP Guidelines
- Regulatory Submission Documents
- Aggregate Safety Reports
- Periodic Safety Update Reports (PSURs)
- Periodic Benefit Risk Evaluation Reports (PBRERs)
- Periodic Adverse Drug Experience Reports (PADERs)
- Development Safety Update Reports (DSURs)
- Nonclinical reports and common technical document (CTD) summary modules
- Integrated summary of safety (ISS), integrated summary of efficacy (ISE) and clinical CTD modules for marketing applications
OTHERS
- Daily Tasks
- Certification Guidelines
- Sample Questions
- CV/Resume Preparation
- Mock Interviews
- Group Discussions
- Personality Development